About BioCor
Lluís Revilla
August Pi i Sunyer Biomedical Research Institute (IDIBAPS); Liver Unit, Hospital Cliniclrevilla@clinic.cat
Juan José Lozano
Centro de Investigación Biomédica en Red de Enfermedades Hepaticas y Digestivas (CIBEREHD); Barcelona, SpainPau Sancho-Bru
August Pi i Sunyer Biomedical Research Institute (IDIBAPS); Liver Unit, Hospital Clinic20 October 2025
Source:vignettes/BioCor_1_basics.Rmd
BioCor_1_basics.RmdAbstract
Describes the background of the package, important functions defined in the package and some of the applications and usages, including the integration with other packages and analysis, and comparisons with related packages. Some frequent or important questions about the package are answered at the end of the document. For more advanced usage you can look at the other vignette.
Introduction
Methods to find similarities have been developed for several purposes, being Jaccard and Dice similarities the most known. In bioinformatics much of the research on the topic is centered around Gene Ontologies because they provide controlled vocabularies, as part of their mission:
The mission of the GO Consortium is to develop an up-to-date, comprehensive, computational model of biological systems, from the molecular level to larger pathways, cellular and organism-level systems.
However, there is another resource of similarities between genes: metabolic pathways. Metabolic pathways describe the relationship between genes, proteins, lipids and other elements of the cells. A pathway describes, to some extent, the function in which it is involved in the cell. There exists several databases about which gene belong to which pathway. Together with pathways, gene sets related to a function or to a phenotype are a source of information of the genes function. With this package we provide the methods to calculate functional similarities based on this information.
Here we provides functions to calculate functional similarities distances for pathways, gene sets, genes and clusters of genes. The name BioCor stands from biological correlation, shortened to BioCor, because as said we look if some genes are in the same pathways or gene sets as other genes.
BioCor is different from GeneOverlap because here we use the Dice index instead of the Jaccard index (although we provide a function to change from one to the other, see this section)and that package only allows to compare pathways but not genes or groups of genes. But GeneOverlap provides some functionalities to plot the similarity scores and provides the associated p-value to the comparison of pathways.
The development of this package aimed initially to improve clustering
of genes by functionality in weighted gene co-expression networks using
WGCNA.
The package has some functions to combine similarities in order to
integrate with WGCNA. For other uses you can check the advanced
vignette..
Installation
The BioCor package is available at Bioconductor and can be downloaded and installed via BiocManager:
install.packages("BiocManager")
BiocManager::install("BioCor")You can install the latest version of BioCor from Github with:
library("devtools")
install_github("llrs/BioCor")Using BioCor
Preparation
We can load the package and prepare the data for which we want to calculate the similarities:
library("BioCor")
## Load libraries with the data of the pathways
library("org.Hs.eg.db")
library("reactome.db")
genesKegg <- as.list(org.Hs.egPATH)
genesReact <- as.list(reactomeEXTID2PATHID)
# Remove genes and pathways which are not from human pathways
genesReact <- lapply(genesReact, function(x){
unique(grep("R-HSA-", x, value = TRUE))
})
genesReact <- genesReact[lengths(genesReact) >= 1] To avoid having biased data it is important to have all the data about the pathways and genes associated to all pathways for the organism under study. Here we assume that we are interested in human pathways. We use this two databases KEGG and Reactome as they are easy to obtain the data. However KEGG database is no longer free for large retrievals therefore it is not longer updated in the Bioconductor annotation packages.
However, one can use any list where the names of the list are the genes and the elements of the list the pathways or groups where the gene belong. One could also read from a GMT file or use GeneSetCollections in addition or instead of those associations from a pathway database and convert it to list using:
library("GSEABase")
paths2Genes <- geneIds(getGmt("/path/to/file.symbol.gmt",
geneIdType=SymbolIdentifier()))
genes <- unlist(paths2Genes, use.names = FALSE)
pathways <- rep(names(paths2Genes), lengths(paths2Genes))
genes2paths <- split(pathways, genes) # List of genes and the gene setsWith genes2paths we have the information ready to
use.
Pathway similarities
We can compute similarities (Dice similarity, see question 1 of FAQ) between two pathways or between several pathways and combine them, or not:
(paths <- sample(unique(unlist(genesReact)), 2))
## [1] "R-HSA-8956320" "R-HSA-75067"
pathSim(paths[1], paths[2], genesReact)
## [1] 0
(pathways <- sample(unique(unlist(genesReact)), 10))
## [1] "R-HSA-5632927" "R-HSA-1839128" "R-HSA-1638091" "R-HSA-9006821"
## [5] "R-HSA-5619088" "R-HSA-3656243" "R-HSA-9022927" "R-HSA-1296346"
## [9] "R-HSA-190241" "R-HSA-9013405"
mpathSim(pathways, genesReact)
## R-HSA-5632927 R-HSA-1839128 R-HSA-1638091 R-HSA-9006821
## R-HSA-5632927 1 0 0 0
## R-HSA-1839128 0 1 0 0
## R-HSA-1638091 0 0 1 0
## R-HSA-9006821 0 0 0 1
## R-HSA-5619088 0 0 0 0
## R-HSA-3656243 0 0 0 0
## R-HSA-9022927 0 0 0 0
## R-HSA-1296346 0 0 0 0
## R-HSA-190241 0 0 0 0
## R-HSA-9013405 0 0 0 0
## R-HSA-5619088 R-HSA-3656243 R-HSA-9022927 R-HSA-1296346
## R-HSA-5632927 0 0 0 0
## R-HSA-1839128 0 0 0 0
## R-HSA-1638091 0 0 0 0
## R-HSA-9006821 0 0 0 0
## R-HSA-5619088 1 0 0 0
## R-HSA-3656243 0 1 0 0
## R-HSA-9022927 0 0 1 0
## R-HSA-1296346 0 0 0 1
## R-HSA-190241 0 0 0 0
## R-HSA-9013405 0 0 0 0
## R-HSA-190241 R-HSA-9013405
## R-HSA-5632927 0 0
## R-HSA-1839128 0 0
## R-HSA-1638091 0 0
## R-HSA-9006821 0 0
## R-HSA-5619088 0 0
## R-HSA-3656243 0 0
## R-HSA-9022927 0 0
## R-HSA-1296346 0 0
## R-HSA-190241 1 0
## R-HSA-9013405 0 1When the method to combine the similarities is set to
NULL mpathSim() returns a matrix of pathway
similarities, otherwise it combines the values. In the next section we
can see the methods to combine pathway similarities.
Combining values
To combine values we provide a function with several methods:
sim <- mpathSim(pathways, genesReact)
methodsCombineScores <- c("avg", "max", "rcmax", "rcmax.avg", "BMA",
"reciprocal")
sapply(methodsCombineScores, BioCor::combineScores, scores = sim)
## avg max rcmax rcmax.avg BMA reciprocal
## 0.1 1.0 1.0 1.0 1.0 1.0We can also specify the method to combine the similarities in
mpathSim(), geneSim(),
mgeneSim(), clusterSim(),
mclusterSim(), clusterGeneSim() and
mclusterGeneSim(), argument method. By default the method
is set to “max” to combine pathways (except in mpathSim where the
default is to show all the pathway similarities) and “BMA” to combine
similarities of genes or for cluster analysis. This function is adapted
from GOSemSim
package.
The function combineScoresPar() allows to use a parallel
background (using BiocParallel)
to combine the scores. It is recommended to use a parallel background if
you calculate more than 300 gene similarities. It also have an argument
in case you want to calculate the similarity scores of several sets.
Gene similarities
To compare the function of two genes there is the
geneSim function and mgeneSim function for
several comparisons. In this example we compare the genes BRCA1 and
BRCA2 and NAT2, which are the genes 672, 675 and 10 respectively in
ENTREZID:
geneSim("672", "675", genesKegg)
## [1] 0.0824295
geneSim("672", "675", genesReact)
## [1] 1
mgeneSim(c("BRCA1" = "672", "BRCA2" = "675", "NAT2" = "10"), genesKegg)
## BRCA1 BRCA2 NAT2
## BRCA1 1.0000000 0.082429501 0.000000000
## BRCA2 0.0824295 1.000000000 0.008241758
## NAT2 0.0000000 0.008241758 1.000000000
mgeneSim(c("BRCA1" = "672", "BRCA2" = "675", "NAT2" = "10"), genesReact)
## BRCA1 BRCA2 NAT2
## BRCA1 1.0000000 1.0000000 0.2322853
## BRCA2 1.0000000 1.0000000 0.2322853
## NAT2 0.2322853 0.2322853 1.0000000Note that for the same genes each database or list provided has different annotations, which result on different similarity scores. In this example BRCA1 has 3 and 27 pathways in KEGG and Reactome respectively and BRCA2 has 1 and 59 pathways in KEGG and Reactome respectively which results on different scores.
Gene cluster similarities
There are two methods:
- Combining all the pathways for each cluster and compare between them.
- Calculate the similarity between genes of a cluster and the other cluster.
By pathways
As explained, in this method all the pathways of a cluster are compared with all the pathways of the other cluster. If a method to combine pathways similarities is not provided, all pathway similarities are returned:
clusterSim(c("672", "675"), c("100", "10", "1"), genesKegg)
## [1] 0.04210526
clusterSim(c("672", "675"), c("100", "10", "1"), genesKegg, NULL)
## 00230 01100 05340 00232 00983
## 04120 0.00000000 0.000000000 0.011764706 0 0
## 03440 0.04210526 0.006908463 0.000000000 0 0
## 05200 0.00000000 0.008241758 0.005540166 0 0
## 05212 0.00000000 0.001666667 0.019047619 0 0
clusters <- list(cluster1 = c("672", "675"),
cluster2 = c("100", "10", "1"),
cluster3 = c("18", "10", "83"))
mclusterSim(clusters, genesKegg, "rcmax.avg")
## cluster1 cluster2 cluster3
## cluster1 1.00000000 0.01587957 0.00256157
## cluster2 0.01587957 1.00000000 0.56412591
## cluster3 0.00256157 0.56412591 1.00000000
mclusterSim(clusters, genesKegg, "max")
## cluster1 cluster2 cluster3
## cluster1 1.000000000 0.04210526 0.008241758
## cluster2 0.042105263 1.00000000 1.000000000
## cluster3 0.008241758 1.00000000 1.000000000By genes
In this method first the similarities between each gene is calculated, then the similarity between each group of genes is calculated. Requiring two methods to combine values, the first one to combine pathways similarities and the second one to combine genes similarities. If only one is provided it returns the matrix of similarities of the genes of each cluster:
clusterGeneSim(c("672", "675"), c("100", "10", "1"), genesKegg)
## [1] 0.02605425
clusterGeneSim(c("672", "675"), c("100", "10", "1"), genesKegg, "max")
## 100 10 1
## 672 0.01176471 0.000000000 NA
## 675 0.04210526 0.008241758 NA
mclusterGeneSim(clusters, genesKegg, c("max", "rcmax.avg"))
## cluster1 cluster2 cluster3
## cluster1 1.000000000 0.02605425 0.006181319
## cluster2 0.026054248 1.00000000 1.000000000
## cluster3 0.006181319 1.00000000 1.000000000
mclusterGeneSim(clusters, genesKegg, c("max", "max"))
## cluster1 cluster2 cluster3
## cluster1 1.000000000 0.04210526 0.008241758
## cluster2 0.042105263 1.00000000 1.000000000
## cluster3 0.008241758 1.00000000 1.000000000Note the differences between mclusterGeneSim() and
mclusterSim() in the similarity values of the clusters. If
we set method = c("max", "max") in
mclusterGeneSim() then the similarity between the clusters
is the same as clusterSim().
Converting similarities
If needed, Jaccard similarity can be calculated from Dice similarity
using D2J():
D2J(sim)
## R-HSA-5632927 R-HSA-1839128 R-HSA-1638091 R-HSA-9006821
## R-HSA-5632927 1 0 0 0
## R-HSA-1839128 0 1 0 0
## R-HSA-1638091 0 0 1 0
## R-HSA-9006821 0 0 0 1
## R-HSA-5619088 0 0 0 0
## R-HSA-3656243 0 0 0 0
## R-HSA-9022927 0 0 0 0
## R-HSA-1296346 0 0 0 0
## R-HSA-190241 0 0 0 0
## R-HSA-9013405 0 0 0 0
## R-HSA-5619088 R-HSA-3656243 R-HSA-9022927 R-HSA-1296346
## R-HSA-5632927 0 0 0 0
## R-HSA-1839128 0 0 0 0
## R-HSA-1638091 0 0 0 0
## R-HSA-9006821 0 0 0 0
## R-HSA-5619088 1 0 0 0
## R-HSA-3656243 0 1 0 0
## R-HSA-9022927 0 0 1 0
## R-HSA-1296346 0 0 0 1
## R-HSA-190241 0 0 0 0
## R-HSA-9013405 0 0 0 0
## R-HSA-190241 R-HSA-9013405
## R-HSA-5632927 0 0
## R-HSA-1839128 0 0
## R-HSA-1638091 0 0
## R-HSA-9006821 0 0
## R-HSA-5619088 0 0
## R-HSA-3656243 0 0
## R-HSA-9022927 0 0
## R-HSA-1296346 0 0
## R-HSA-190241 1 0
## R-HSA-9013405 0 1Also if one has a Jaccard similarity and wants a Dice similarity, can
use the J2D() function.
High volumes of gene similarities
We can compute the whole similarity of genes in KEGG or Reactome by using :
## Omit those genes without a pathway
nas <- sapply(genesKegg, function(y){all(is.na(y)) | is.null(y)})
genesKegg2 <- genesKegg[!nas]
m <- mgeneSim(names(genesKegg2), genesKegg2, method = "max")It takes around 5 hours in one core but it requires high memory available.
If one doesn’t have such a memory available can compute the similarities by pieces, and then fit it in another matrix with:
sim <- AintoB(m, B)Usually B is a matrix of size length(genes), see
?AintoB().
An example of usage
In this example I show how to use BioCor to analyse a list of genes by functionality. With a list of genes we are going to see how similar are those genes:
genes.id <- c("10", "15", "16", "18", "2", "9", "52", "3855", "3880", "644",
"81327", "9128", "2073", "2893", "5142", "60", "210", "81",
"1352", "88", "672", "675")
genes.id <- mapIds(org.Hs.eg.db, keys = genes.id, keytype = "ENTREZID",
column = "SYMBOL")
## 'select()' returned 1:1 mapping between keys and columns
genes <- names(genes.id)
names(genes) <- genes.id
react <- mgeneSim(genes, genesReact)
## Warning in mgeneSim(genes, genesReact): Some genes are not in the list
## provided.
## We remove genes which are not in list (hence the warning):
nan <- genes %in% names(genesReact)
react <- react[nan, nan]
hc <- hclust(as.dist(1 - react))
plot(hc, main = "Similarities between genes")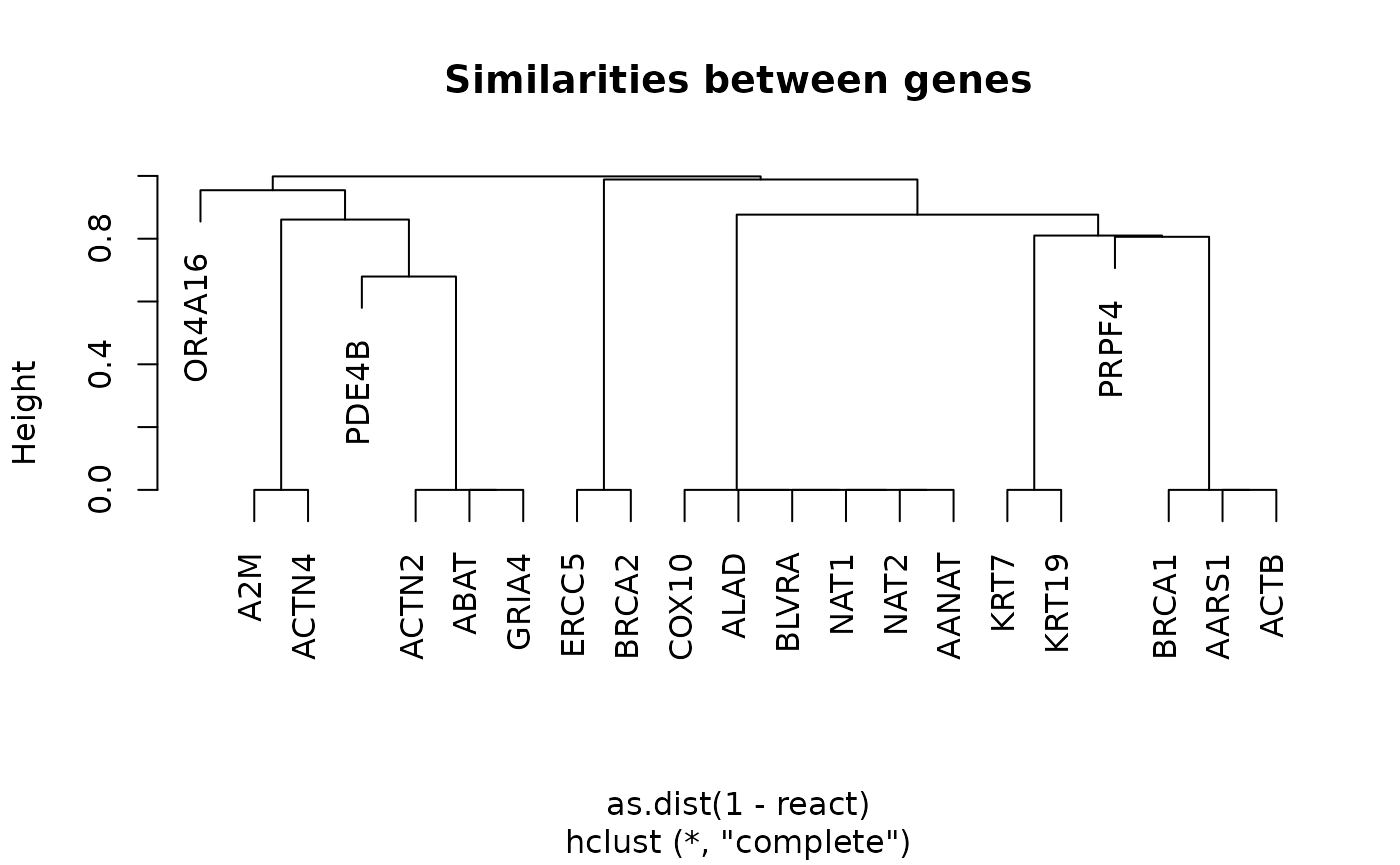
Gene clustering by similarities
Now we can see the relationship between the genes. We can group them for a cluster analysis to visualize the relationship between the clusters:
mycl <- cutree(hc, h = 0.2)
clusters <- split(genes[nan], as.factor(mycl))
# Removing clusters of just one gene
(clusters <- clusters[lengths(clusters) >= 2])
## $`1`
## NAT2 AANAT NAT1 BLVRA ALAD COX10
## "10" "15" "9" "644" "210" "1352"
##
## $`2`
## AARS1 ACTB BRCA1
## "16" "60" "672"
##
## $`3`
## ABAT GRIA4 ACTN2
## "18" "2893" "88"
##
## $`4`
## A2M ACTN4
## "2" "81"
##
## $`5`
## KRT7 KRT19
## "3855" "3880"
##
## $`8`
## ERCC5 BRCA2
## "2073" "675"
names(clusters) <- paste0("cluster", names(clusters))
## Remember we can use two methods to compare clusters
sim_clus1 <- mclusterSim(clusters, genesReact)
plot(hclust(as.dist(1 - sim_clus1)),
main = "Similarities between clusters by pathways")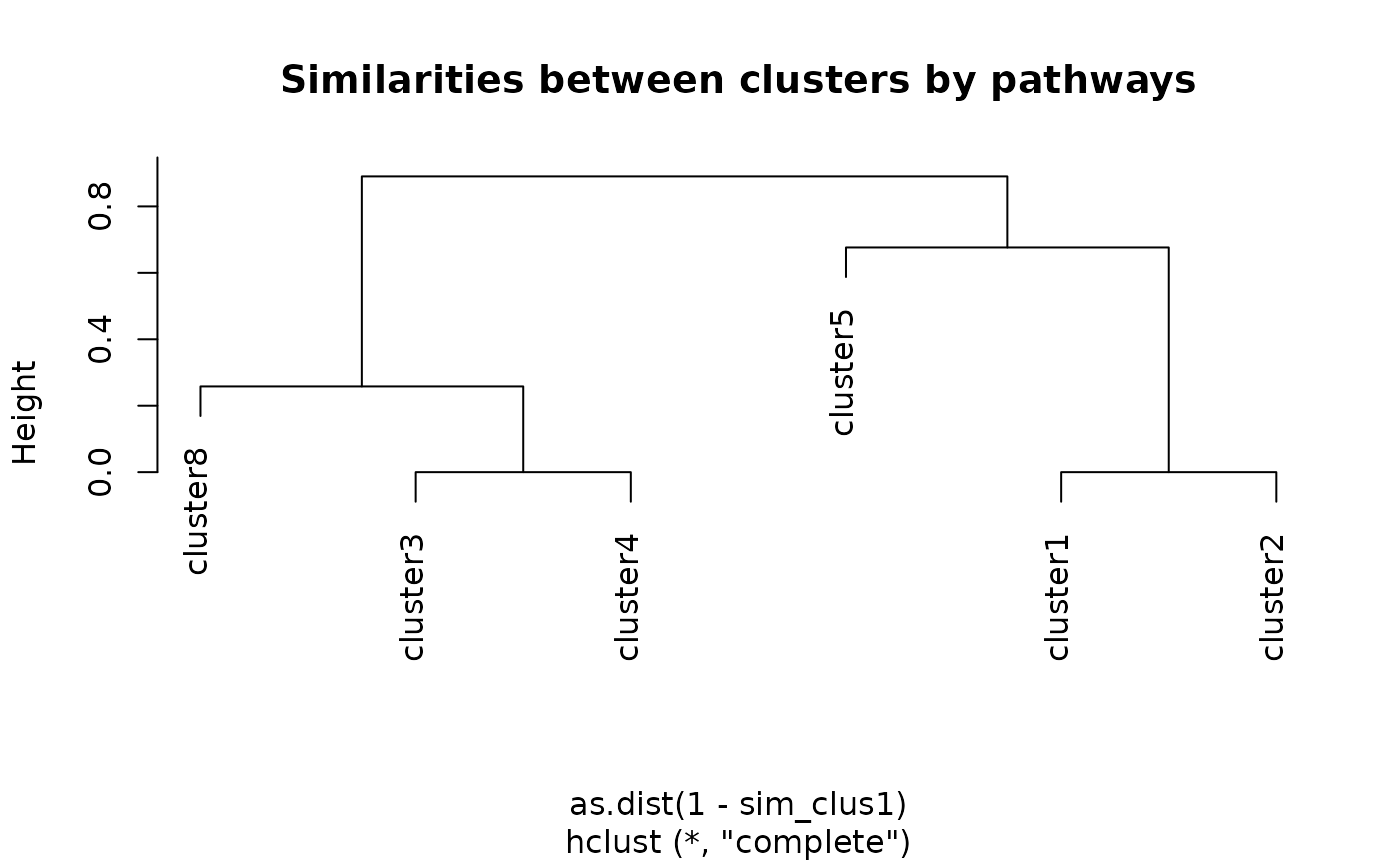
Clustering using clusterSim
sim_clus2 <- mclusterGeneSim(clusters, genesReact)
plot(hclust(as.dist(1 - sim_clus2)),
main ="Similarities between clusters by genes")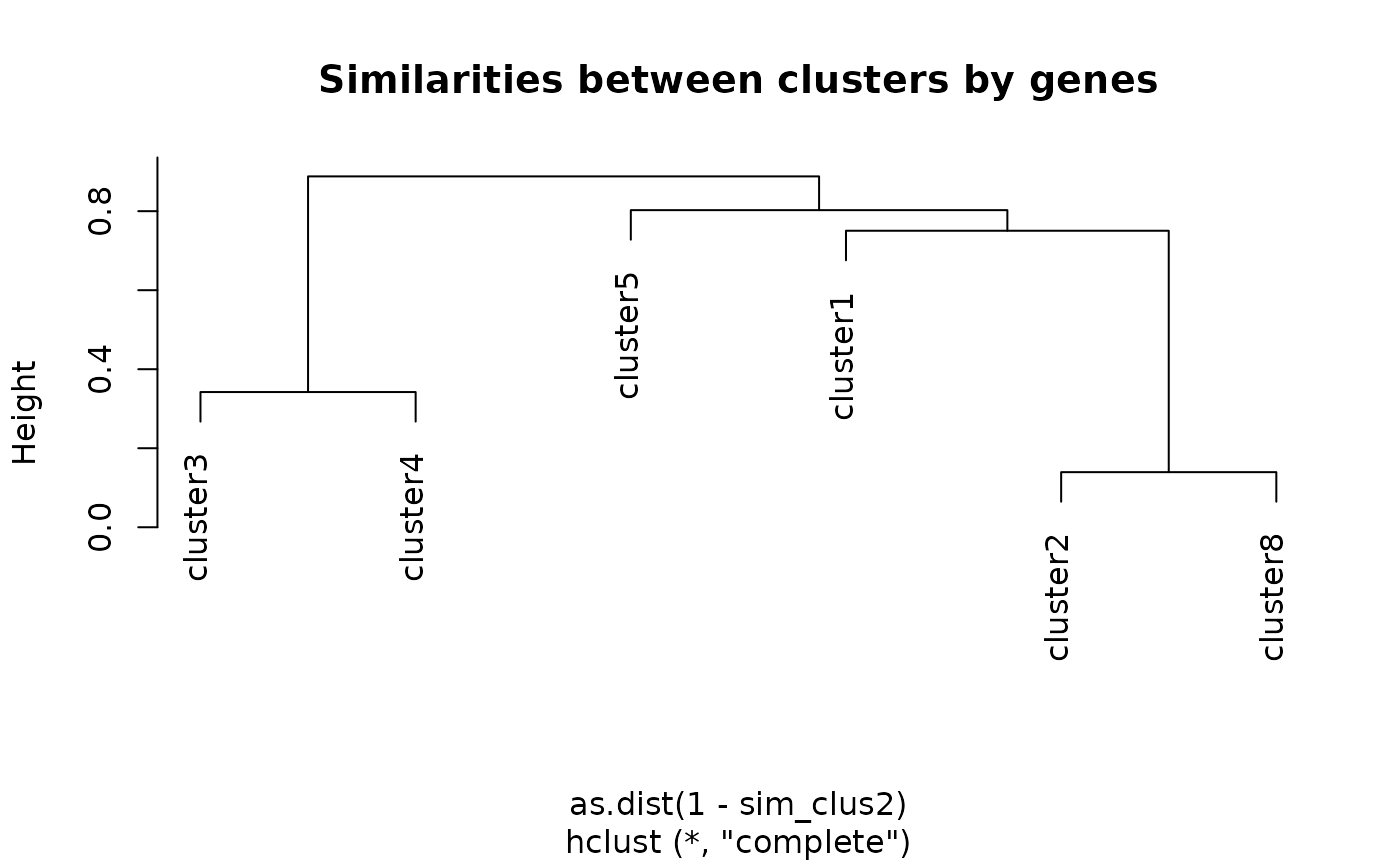
Clustering using clusterGeneSim
Each method results in a different dendrogram as we can see on Figure @ref(fig:hclust3) compared to Figure @ref(fig:hclust3b).
Comparing with GOSemSim
In this section I will compare the functional similarity of BioCor with the closely related package GOSemSim. The genes and gene clusters used were extracted from GOSemSim’s vignette, we only change the ontology, instead of the molecular function, the biological process will be used:
hsGO <- GOSemSim::godata('org.Hs.eg.db', ont = "BP", computeIC = FALSE)
##
## Warning in GOSemSim::godata("org.Hs.eg.db", ont = "BP", computeIC = FALSE): use
## 'annoDb' instead of 'OrgDb'
## preparing gene to GO mapping data...I will compare the functions geneSim from section geneSim and mgeneSim from GOSemSim with both data sets from KEGG and Reactome:
goSemSim <- GOSemSim::geneSim("241", "251", semData = hsGO,
measure = "Wang", combine="BMA")
# In case it is null
sim <- ifelse(is.na(goSemSim), 0, getElement(goSemSim, "geneSim"))
BioCor::geneSim("241", "251", genesReact, "BMA") - sim
## [1] 0.04844925
genes <- c("835", "5261","241", "994")
goSemSim <- GOSemSim::mgeneSim(genes, semData = hsGO,
measure = "Wang", combine = "BMA",
verbose = FALSE, drop = NULL)
BioCor::mgeneSim(genes, genesReact, "BMA", round = TRUE) - goSemSim
## 835 5261 241 994
## 835 0.000 -0.266 -0.138 -0.148
## 5261 -0.266 0.000 0.030 -0.271
## 241 -0.138 0.030 0.000 -0.119
## 994 -0.148 -0.271 -0.119 0.000We can observe there is more similarity according to the gene
ontology than according to the pathways. See FAQ question 8 about the use of BioCor::
and GOSemSim::.
If named characters are passed they are used to name the resulting matrix:
genes <- c("CDC45", "MCM10", "CDC20", "NMU", "MMP1")
genese <- mapIds(org.Hs.eg.db, keys = genes, column = "ENTREZID",
keytype = "SYMBOL")
## 'select()' returned 1:1 mapping between keys and columns
BioCor::mgeneSim(genese, genesReact, "BMA")
## CDC45 MCM10 CDC20 NMU MMP1
## CDC45 1.00000000 0.90725461 0.4863010 0.04975896 0.08559666
## MCM10 0.90725461 1.00000000 0.4685054 0.05888261 0.09179822
## CDC20 0.48630097 0.46850540 1.0000000 0.15897580 0.21571940
## NMU 0.04975896 0.05888261 0.1589758 1.00000000 0.11570387
## MMP1 0.08559666 0.09179822 0.2157194 0.11570387 1.00000000We can further compare the cluster similarities from the next section of the vignette:
gs1 <- c("835", "5261","241", "994", "514", "533")
gs2 <- c("578","582", "400", "409", "411")
BioCor::clusterSim(gs1, gs2, genesReact, "BMA") -
GOSemSim::clusterSim(gs1, gs2, hsGO, measure = "Wang", combine = "BMA")
## [1] -0.184594
x <- org.Hs.egGO
hsEG <- mappedkeys(x)
set.seed(123)
(clusters <- list(a=sample(hsEG, 20), b=sample(hsEG, 20), c=sample(hsEG, 20)))
## $a
## [1] "5267" "9764" "4538" "2803" "5104" "57799" "6993" "10092"
## [9] "284004" "4097" "80179" "28773" "54505" "90423" "4288" "9350"
## [17] "23281" "51071" "79867" "90324"
##
## $b
## [1] "2334" "390437" "117143" "153743" "29110" "51637" "28564" "160365"
## [9] "442919" "4594" "152217" "23209" "5939" "85437" "25759" "349565"
## [17] "55623" "9410" "693182" "198437"
##
## $c
## [1] "102724127" "79740" "84066" "375033" "25936" "91750"
## [7] "53" "125981" "10940" "10788" "256957" "55884"
## [13] "9703" "10085" "57506" "26094" "51719" "54674"
## [19] "79541" "100422911"
BioCor::mclusterSim(clusters, genesReact, "BMA") -
GOSemSim::mclusterSim(clusters, hsGO, measure = "Wang", combine = "BMA")
## Warning in BioCor::mclusterSim(clusters, genesReact, "BMA"): Some genes are not
## in the list provided.
## a b c
## a 0.00000000 0.041358282 0.106733541
## b 0.04135828 0.000000000 -0.007613623
## c 0.10673354 -0.007613623 0.000000000WGCNA and BioCor
WGCNA uses the correlation of the expression data of several samples to cluster genes. Sometimes, from a biological point of view the interpretation of the resulting modules is difficult, even more when some groups of genes end up not having an enrichment in previously described functions. BioCor was originally thought to be used to overcome this problem: to help clustering genes, not only by correlation but also by functionality.
In order to have groups functionally related, functional similarities can enhance the clustering of genes when combined with experimental correlations. The resulting groups will reflect, not only the correlation of the expression provided, but also the functionality known of those genes.
We propose the following steps:
- Calculate the similarities for the expression data
- Calculate the similarities of the genes in the expression
- Combine the similarities
- Calculate the adjacency
- Identify modules with hierarchical clustering
Here we provide an example on how to use BioCor with WGCNA:
sim is a list where each element is a matrix of
similarities between genes Our normalized expression is in the
expr variable, a matrix where the samples are in the rows
and genes in the columns.
expr.sim <- WGCNA::cor(expr) # or bicor
## Combine the similarities
similarity <- similarities(c(list(exp = expr.sim), sim), mean, na.rm = TRUE)
## Choose the softThreshold
pSFT <- pickSoftThreshold.fromSimilarity(similarity)
## Or any other function we want
adjacency <- adjacency.fromSimilarity(similarity, power = pSFT$powerEstimate)
## Once we have the similarities we can calculate the TOM with TOM
TOM <- TOMsimilarity(adjacency) ## Requires adjacencies despite its name
dissTOM <- 1 - TOM
geneTree <- hclust(as.dist(dissTOM), method = "average")
## We can use a clustering tool to group the genes
dynamicMods <- cutreeHybrid(dendro = geneTree, distM = dissTOM,
deepSplit = 2, pamRespectsDendro = FALSE,
minClusterSize = 30)
moduleColors <- labels2colors(dynamicMods$labels)Once the modules are identified using the functional similarities of this package and the gene correlations, one can continue with the workflow of WGCNA.
An important aspect in this process is deciding how to combine the similarities and the expression data: - If the functional similarities play a huge role, we will end up having only those genes closely related to the same functions. - If the functional similarities play a low role, it will be similarly to only use WGCNA, and the genes won’t be functionally related.
For these reasons it is better to use weights between
0.5 and 1 for expression if you use
weighted.sum or similar functions.
There are several things to take into account when choosing a way to combine: - The size of the gray or 0 modules (those who don’t show a specific pattern) - The number and size of the modules created. - The way the similarities are combined
Violin plots may help to view the differences in size and distribution of the modules across different methods of combining the similarities.
FAQ
How is defined the pathway similarity?
BioCor uses the Sørensen–Dice index: The dice similarity is the double of the genes shared by the pathways divided by the number of genes in each pathway.
We can calculate the similarity between two pathways (, ) with:
This is implemented in the diceSim function, which
results is similar to Jaccard index:
Both Jaccard index and dice index are between 0 and 1
().
To calculate the Jaccard index from the diceSim use the
D2J function.
Why does BioCor use the dice coefficient and not the Jaccard ?
We consider Dice coefficient better than Jaccard because it has higher values for the same comparisons, which reflects that including a gene in a pathway is not easily done.
How does BioCor combine similarities between several pathways of two genes?
Although the recommend method is the “max” method, (set as default),
there are implemented other methods in combineScores of the
GOSemSim
package which I borrowed1.
Why do you recommend using the max method to combine similarities scores for pathways?
The purpose of combining the scores is usually to find the relationships between genes through their pathways. The higher the similarity is between two pathway of two genes, the higher functionality do the genes share, even if those genes have other non-related functions.
How to detect which functional relationship is more important between two genes?
If two genes are involved in the same pathways usually they have (to some extent, maybe indirect) interactions. To detect which relationship is more important between two genes one could measure other similarities scores and check the stoichiometry of the pathways and measure the expression changes and correlation between them or use dynamic simulations of the pathways.
How to detect with which genes is my gene of interest related?
You can measure the gene similarity between those genes and also measure the expression correlation of your gene of interest with other genes.
Why isn’t available a method for calculating GO similarities?
This is covered by the GOSemSim
package, you can use it to produce a similarity matrix (i.e. use
mgeneSim). You can parallelize it with foreach
package or BiocParallel if your list of genes is big.
I get an error! How do I solve this?
If the error is like this:
Error in FUN(X[[i]], ...) :
trying to get slot "geneAnno" from an object of a basic class ("list") with no slotsAnd you have loaded the GOSemSim library, R is calling
the GOSemSim function of the same name. Use BioCor:: to
call the function from BioCor (f.ex:
BioCor::geneSim)
If the error is not previously described in the support forum, post a question there.
My apologies if you found a bug or an inconsistency between what
BioCor should do and what it actually does. Once you
checked that it is a bug, please let me know at the issues page of
Github.
Session Info
sessionInfo()
## R version 4.5.1 (2025-06-13)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] reactome.db_1.92.0 org.Hs.eg.db_3.21.0 AnnotationDbi_1.70.0
## [4] IRanges_2.42.0 S4Vectors_0.46.0 Biobase_2.68.0
## [7] BiocGenerics_0.54.1 generics_0.1.4 BioCor_1.33.3
## [10] BiocStyle_2.36.0
##
## loaded via a namespace (and not attached):
## [1] KEGGREST_1.48.1 GOSemSim_2.34.0 xfun_0.53
## [4] bslib_0.9.0 htmlwidgets_1.6.4 lattice_0.22-7
## [7] yulab.utils_0.2.1 vctrs_0.6.5 tools_4.5.1
## [10] parallel_4.5.1 RSQLite_2.4.3 blob_1.2.4
## [13] R.oo_1.27.1 pkgconfig_2.0.3 Matrix_1.7-3
## [16] desc_1.4.3 graph_1.86.0 lifecycle_1.0.4
## [19] GenomeInfoDbData_1.2.14 compiler_4.5.1 textshaping_1.0.4
## [22] Biostrings_2.76.0 codetools_0.2-20 GenomeInfoDb_1.44.3
## [25] htmltools_0.5.8.1 sass_0.4.10 yaml_2.3.10
## [28] pkgdown_2.1.3 crayon_1.5.3 jquerylib_0.1.4
## [31] GO.db_3.21.0 R.utils_2.13.0 BiocParallel_1.42.2
## [34] cachem_1.1.0 digest_0.6.37 bookdown_0.45
## [37] fastmap_1.2.0 grid_4.5.1 cli_3.6.5
## [40] XML_3.99-0.19 GSEABase_1.70.1 rappdirs_0.3.3
## [43] UCSC.utils_1.4.0 bit64_4.6.0-1 rmarkdown_2.30
## [46] XVector_0.48.0 httr_1.4.7 bit_4.6.0
## [49] ragg_1.5.0 png_0.1-8 R.methodsS3_1.8.2
## [52] memoise_2.0.1 evaluate_1.0.5 knitr_1.50
## [55] rlang_1.1.6 xtable_1.8-4 DBI_1.2.3
## [58] BiocManager_1.30.26 annotate_1.86.1 jsonlite_2.0.0
## [61] R6_2.6.1 systemfonts_1.3.1 fs_1.6.6